{kind=link}
Блог пользователя mordovtseva
Новые подробности патогенеза и терапии опухолей при туберозном склерозе
сб., 21/12/2019 - 20:42 — mordovtsevaПродолжаются исследования патогенеза туберозного склероза, уточняются его фенотипические особенности. За последние пять лет был достигнут значительный прогресс в понимании патогенеза опухолей на молекулярно-генетическом уровне, в том числе благодаря методам секвенирования нового поколения.
Известно, что ангиофибромы, состоящие из фибробластоподобных клеток, волокнистой ткани, сосудов и волосяных фолликулов, являются одним из наиболее характерных клинических признаков туберозного склероза. Гены в клеточных элементах ангиофибром, в частности, в фибробластоподобных клетках, часто подвергаются повторным мутациям. Предполагается, что последние происходят из мезенцефального нервного гребня, что объясняет превалирующую локализацию ангиофибром на лице. Повторные мутации могут быть в виде делеции, инверсии или носить точечный характер. Интересно, что 50% всех соматических точечных мутаций являются результатом повреждения ДНК УФ излучением.
Туберозный склероз: пересмотрены критерии постановки диагноза
пт., 13/12/2019 - 11:39 — mordovtsevaТуберозный склероз – редкое заболевание с аутосомно-доминантным типом наследования и выраженной фенотипической гетерогенностью. В основе заболевания лежат мутации гена TSC1, локализованного на хромосоме 9q34 и кодирующего белок гамартин, или гена TSC2 на хромосоме 16q13, кодирующего белок туберин. Гамартин подавляет рост опухолей, а туберин регулирует эндоцитоз. В некоторых случаях имеет место дефект репарации ДНК под воздействием ионизирующего излучения. При наличии мутации TSC1, что наблюдается приблизительно у 20% пациентов, заболевание протекает в более легкой форме. Только треть случаев – имеют семейную историю, все остальные – результат новых мутаций.
Акродерматит энтеропатический: новые возможности терапии
вс., 08/12/2019 - 18:53 — mordovtsevaЕдинственным методом лечения пациентов до настоящего времени было пожизненное применение препаратов цинка, которое давало хорошие клинические результаты. Однако, в связи с тем, что эти лекарственные средства являлись высокодозированными, то они часто приводили к развитию побочных эффектов (в первую очередь со стороны желудочно-кишечного тракта) и способствовали нарушениям концентрации в сыворотке крови других микроэлементов, в частности, меди и железа, а также иммунному дисбалансу.
Гетерогенные патологические состояния, связанные с недостаточностью цинка
чт., 05/12/2019 - 23:02 — mordovtsevaВыявлены следующие причины недостаточности цинка в организме человека:
А. Мутации в гене SLC39A4, локализованного на хромосоме 8q24.3 и кодирующего протеин Zip4. Данный протеин необходим для всасывания цинка в двенадцатиперстной и тощей кишке. Эти мутации и приводят к развитию энтеропатического акродерматита – наиболее тяжелой формы недостаточности цинка. Как правило, энтеропатический акродерматит проявляется клинически в раннем постнатальном периоде в виде эритематозных, везикулобуллезных очагов на коже, кишечными расстройствами (в первую очередь диареей), выпадением волос. К другим часто встречающимся симптомам заболевания относятся потеря веса, замедление роста, паронихия, ониходистрофия, ангулярный стоматит, хейлит, конъюнктивит, фотофобия, гипогонадизм, анемия. Без адекватной терапии заболевание неуклонно прогрессирует и приводит к гибели больных.
Генодерматозы. Эктодермальные дисплазии и коморбидные состояния
вс., 24/11/2019 - 11:42 — mordovtseva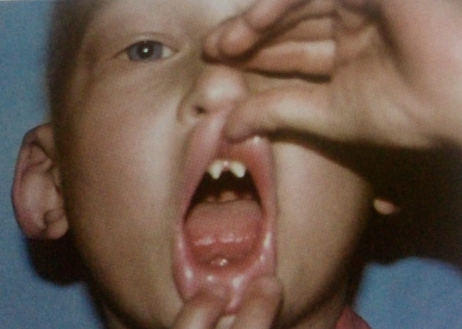 Эктодермальные дисплазии – это группа синдромов, при которых наблюдается поражение тканей, развивающихся из эмбриональной эктодермы. Эта группа включает в себя более 200 клинических форм, которые отличаются уникальными сочетаниями эктодермальных аномалий. Такая уникальность приводит к значительным проблемам классификации, которые пытались решить участники двух международных конференций в 2008 и 2013 годах, но пока не очень успешно. У всех этих редких состояний имеются общие черты, а именно: нарушения в развитии волос, зубов, ногтей и потовых желез. Однако при большинстве заболеваний из этой группы также имеет место патология других органов и систем. Для некоторых из них в настоящее время известны мутации генов, запускающие патологический процесс.
Эктодермальные дисплазии – это группа синдромов, при которых наблюдается поражение тканей, развивающихся из эмбриональной эктодермы. Эта группа включает в себя более 200 клинических форм, которые отличаются уникальными сочетаниями эктодермальных аномалий. Такая уникальность приводит к значительным проблемам классификации, которые пытались решить участники двух международных конференций в 2008 и 2013 годах, но пока не очень успешно. У всех этих редких состояний имеются общие черты, а именно: нарушения в развитии волос, зубов, ногтей и потовых желез. Однако при большинстве заболеваний из этой группы также имеет место патология других органов и систем. Для некоторых из них в настоящее время известны мутации генов, запускающие патологический процесс.
Генодерматозы. Снова о том, что стоит за красотой и породой братьев наших меньших
вс., 10/11/2019 - 19:25 — mordovtsevaВ предыдущем материале речь шла о том, к каким изменениям могут приводить мутации в генах человека, воспроизведенные у животных, на примере патологии пигментации. Но помимо пигментных изменений следует упомянуть также и о патологии волос.
Одним из примеров являются так называемые шерстистые волосы (wooly hair), наличие которых часто сочетается с гипотрихозом. Известно, что кератины являются основными структурными компонентами волосяных фолликулов, и во внутреннем корневом влагалище локализован 2 тип кератинов 71, 72, 73 и 74. Мутации в гене, кодирующем кератины 71 и 74, приводят к развитию гипотрихоза и патологии стрежней в виде шерстистых волос.
Генодерматозы. О единстве всего живого, или что стоит за красотой и породой
пн., 04/11/2019 - 10:43 — mordovtsevaВ основе биологического сходства всех живых существ, как известно, лежит структурное (клетка), генетическое (молекула ДНК) и биохимическое (классы органических соединений) единство. А факт универсальности биологических законов и механизмов для всего животного мира имеет важное значение для различных отраслей медицины.
В течение столетий человек разводил тех или иных животных за их особенные характеристики. В последнее время животных разводят или создают новые породы в основном из-за их внешнего вида.
В соответствии с идеей единства всего живого на Земле ученые провели интересную параллель между рядом генодерматозов у людей и домашних животных. По мнению авторов, мутации, наблюдающиеся при наследственных заболеваниях у человека, имеют место или могут быть воспроизведены и у животных. При этом у животных фенотипические признаки этих мутаций в некоторых случаях даже более выражены.
Генодерматозы: новые гены и формы кожно-глазного альбинизма
чт., 24/10/2019 - 20:19 — mordovtsevaКожно-глазной альбинизм - это редкое врожденное заболевание с аутосомно-рецессивным типом наследования. Характеризуется гипопигментацией кожи, волос и радужной оболочки глаз различной степени выраженности, а также спектром офтальмологической патологии (нистагм, страбизм, снижение остроты зрения, фотофобия и др.). Заболевание гетерогенное, с неоднородной генетической основой, которую удалось уточнить с помощью методов секвенирования нового поколения. Однако примерно у 20% больных с фенотипом альбинизма генетический дефект до сих пор не известен. В настоящее время такие термины, как тирозиназо-положительный или тирозиназо-отрицательный альбинизм считаются устаревшими.
Генодерматозы: онкологическая настороженность при кожно-глазном альбинизме
вс., 13/10/2019 - 10:35 — mordovtsevaОдним из наиболее часто встречающихся врожденных нарушений пигментации кожи является кожно-глазной альбинизм. В основе заболевания лежит генетически обусловленное нарушение дифференцировки меланоцитов с аутосомно-рецессивным типом наследования. Эта патология может реализоваться на этапе биосинтеза меланина, быть связана с дефектом биогенеза меланосом или их функции, или с нарушением регуляции внутриклеточного транспорта и локализации белков, наобходимых для выработки меланина. В настоящее время известны по крайней мере 4 гена, ответственные за развитие 7 клинических типов (OCA 1-7) кожно-глазного альбинизма: TYR, OCA2, TYRP1, SCL45A2.
Степень пигментации кожи человека индивидуальна и определяется многими факторами: количеством и митотической активностью меланоцитов базального слоя эпидермиса, меланогенной активностью меланосом, их количеством, размером и характером распределения, типом меланина и др. Считается, что степень пигментации кожи, обусловленная меланином (на цвет кожи влияют и другие факторы), обратно пропорциональна риску развития УФ-индуцированных опухолей кожи. Именно транспорт меланина в кератиноциты и формирование перинуклеарной защитной оболочки предохраняют ДНК эпителиальных клеток от ультрафиолетового повреждения.
| © Дерматология в России, 2007-2024
|
О сайте | Правовая информация | |
| Материалы сайта предназначены только для врачей. По вопросам, касающимся Вашего здоровья, обратитесь к специалисту. 18+ | |||